Inspired by a patient. Images obtained at 19 months. T2 above and FLAIR below.

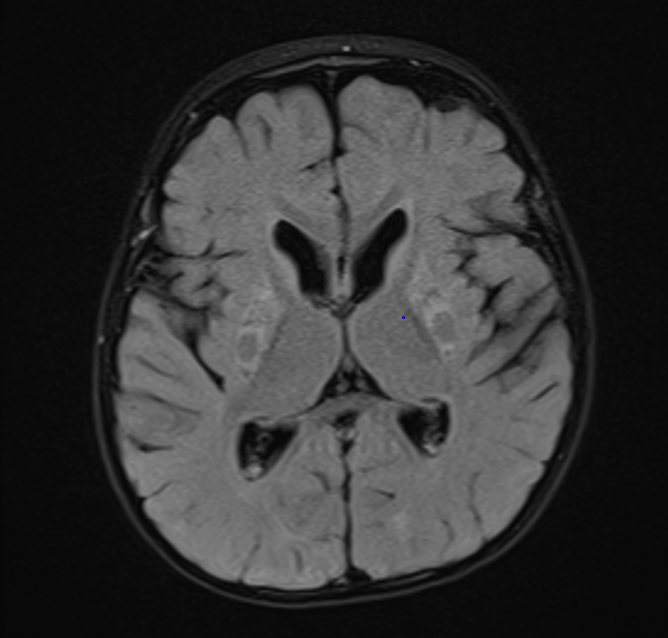
Bhanudeep S, Koneti BB. Pathognomonic Neuroimaging in MEGDEL Syndrome. Indian J Pediatr. 2024 Dec 20. doi: 10.1007/s12098-024-05377-7. Epub ahead of print. PMID: 39704917. (no abstract)

Altamimi R, Aldhalaan H, Tous E, Nicolas-Jilwan M. Teaching NeuroImage: The Putaminal Eye: A Highly Characteristic Imaging Feature of MEGDEL Syndrome. Neurology. 2023 Nov 7;101(19):e1943-e1944. doi: 10.1212/WNL.0000000000207823. Epub 2023 Aug 21. PMID: 37604663; PMCID: PMC10663015. (no abstract)
Monteiro-Cardoso VF, Giordano F. New roles of LPGAT1: From mitochondrial import of phosphatidylglycerol to MEGDEL disease. Cell Rep. 2023 Nov 28;42(11):113376. doi: 10.1016/j.celrep.2023.113376. Epub 2023 Nov 2. PMID: 37917588.
Abstract
Dysregulation of mitochondrial lipidome is associated with several human pathologies. Sun et al.1 show that LPGAT1 cooperates with TIMM14 to regulate phosphatidylglycerol transport from the endoplasmic reticulum to the mitochondria, and uncover the involvement of LPGAT1 deficiency in MEGDEL syndrome.
Alfaraidi AT, ALSulimani NK, Garout W. Incidental Finding of MEGDEL Syndrome at a Tertiary Care Center in Saudi Arabia. Cureus. 2024 Mar 1;16(3):e55308. doi: 10.7759/cureus.55308. PMID: 38559521; PMCID: PMC10981795.
Abstract
MEGDEL syndrome, a rare autosomal recessive disorder characterized by 3-methylglutaconic aciduria, deafness, encephalopathy, and Leigh-like syndrome, results from mutations in the SERAC1 gene. This case report explores the clinical presentation, diagnostic challenges, and genetic findings of an 11-year-old boy with MEGDEL syndrome at a tertiary care center in Saudi Arabia. The patient, born to consanguineous parents, presented with developmental delay, cerebral palsy, intellectual disability, and seizures. Diagnostic evaluation at 15 months revealed 3-methylglutaconic aciduria, and subsequent genetic testing through whole exome sequencing confirmed a rare homozygous deletion variant in the SERAC1 gene. The patient exhibited brain atrophy, tracheal stenosis, laryngomalacia, and skeletal abnormalities. The complexity of MEGDEL syndrome manifestations and the challenge of distinguishing it from other metabolic disorders are discussed, emphasizing the significance of genetic testing in confirming the diagnosis. This case underscores the occurrence of MEGDEL syndrome in a child with cerebral palsy, highlighting the importance of a multidisciplinary approach for diagnosis and the need for genetic counseling in consanguineous families. Although the management remains primarily supportive, the report calls for more comprehensive epidemiological studies to determine the prevalence and incidence of MEGDEL syndrome. The findings contribute to the growing understanding of this rare disorder, thus emphasizing the necessity for ongoing research to enhance diagnostic accuracy and management strategies.
Martins E, Durães J, Nogueira C, Gomes J, Vilarinho L, Macário C. SERAC1 Deficiency- A New Phenotype. Endocr Metab Immune Disord Drug Targets. 2023 Sep 14. doi: 10.2174/1871530323666230914114456. Epub ahead of print. PMID: 37711114.
Abstract
Introduction - SERAC1 deficiency phenotype range from MEGD(H)EL syndrome, the most severe, to juvenile complicated spastic paraplegia, to adult-onset dystonic features (in only one patient). The MEGD(H)EL syndrome is characterized by (3-methylglutaconic aciduria with deafness-dystonia, [hepatopathy], encephalopathy, and Leigh-like syndrome). Biochemical abnormalities: elevated urinary 3 - metilglutaconic and 3-metilglutaric acids, high lactate and alanine in serum. Diagnosis is confirmed when biallelic pathogenic variants in SERAC1 gene are found. Brain MRI: basal ganglia lesions and generalized atrophy. Results/Case report - A 30-year-old patient with a moderate intellectual disability, developed, since the age of 25, a progressive loss of previous capacities (hand dexterity, oral language), and later subacute generalized dystonic features. Currently he has spastic tetraparesis, dystonia, scoliosis and autistic behavior, with bilateral basal ganglia lesions on brain MRI. Genetic study revealed biallelic pathogenic variants in SERAC1 gene, confirm MEGD(H)EL. A 73 years old patient with cognitive impairment and progressive spastic tetraparesis had multiple periventricular T2 hyperintense lesions. She has a homozygotic SERAC1 variant NM_032861: exon4:c.T139A: p.F471 (rs112780453), considered benign. Biochemical study revealed elevated plasmatic alanine and urinary3-metilglutaconic and 3-metilglutaric acid. This profile is concordant with mitochondrial dysfunction and SERAC1 Deficit. Conclusion - The first patient has the clinical symptoms associated to the MEGD(H)EL syndrome, and the biochemical and genetic confirmation of the diagnosis, without reservations. However, in the second patient, the progressive paraparesis and cognitive impairment did not appear to be caused by multiple sclerosis nor subcortical vascular leukoencephalopathy (without vascular risk factors). The abnormal biochemical profile is suggestive of SERAC1 Deficiency, even without genetic confirmation. In what should we believe?
No comments:
Post a Comment